
OGSIVEO 100 mg, comprimés pelliculés, boîte de 1 flacon de 75
Dernière révision : 24/01/2025
Taux de TVA : 0%
Laboratoire exploitant : CEVIDRA
Source :

OGSIVEO est indiqué dans le traitement des patients adultes présentant des tumeurs desmoïdes (TD) en progression après au moins une ligne de traitement antérieure incluant les inhibiteurs de la tyrosine kinase (ITK).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Grossesse (voir rubrique Fertilité, grossesse et allaitement)
Femmes en âge de procréer n'utilisant pas une méthode de contraception hautement efficace (voir rubriques Mises en garde spéciales et précautions d'emploi et Fertilité, grossesse et allaitement)
Allaitement (voir rubrique Fertilité, grossesse et allaitement)
Diarrhée
Une diarrhée a été rapportée chez des patients recevant du nirogacestat (voir rubrique Effets indésirables). Les patients qui présentent une diarrhée pendant le traitement par OGSIVEO doivent être surveillés et pris en chargeà l'aide de médicaments antidiarrhéiques. Pour une diarrhée de grade 3 qui persiste pendant ≥ 3 jours malgré un traitement médical maximal, OGSIVEO doit être suspendu jusqu'à ce que la diarrhée soit revenue à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour (voir rubrique Posologie et mode d'administration).
Affections de la peau et du tissu sous-cutané
Des réactions dermatologiques, incluant rash maculopapuleux, folliculite et hidradénite, ont été rapportées chez des patients recevant du nirogacestat (voir rubrique Effets indésirables). Les patients doivent être surveillés afin de détecter d'éventuelles réactions dermatologiques tout au long du traitement et doivent être pris en charge en fonction du tableau clinique. Pour les réactions dermatologiques de grade 3, OGSIVEO doit être suspendu jusqu'à retour à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour (voir rubrique Posologie et mode d'administration).
Toxicité ovarienne
Une toxicité ovarienne a été rapportée chez des patientes en âge de procréer recevant du nirogacestat (voir rubrique Effets indésirables). Une toxicité ovarienne, identifiée sur la base des taux anormaux d'hormones de reproduction et/ou des symptômes périménopausiques, a été rapportée chez 75 % des femmes en âge de procréer recevant du nirogacestat dans l'étude pivotale. La toxicité ovarienne a disparu chez 79% des femmes en âge de procréer pendant le traitement. Des informations de suivi sont disponibles pour toutes les patientes sauf deux sur 27 ; après l'arrêt du traitement, la toxicité ovarienne a disparu chez toutes les femmes en âge de procréer pour lesquelles des données sont disponibles (voir section Effets indésirables). Les effets du nirogacestat sur la fertilité humaine sont inconnus. D'après les résultats d'études animales, la fertilité féminine peut être altérée. Les femmes en âge de procréer doivent être informées du risque de toxicité ovarienne avant d'entamer un traitement par OGSIVEO. Les patientes doivent être surveillées en cas de modification de la régularité du cycle menstruel ou del'apparition de symptômes de carence en oestrogènes, notamment bouffées de chaleur, sueurs nocturnes et sécheresse vaginale.
Anomalies électrolytiques
Des anomalies électrolytiques, notamment une hypophosphatémie et une hypokaliémie, sont survenues chez des patients recevant du nirogacestat (voir rubrique Effets indésirables). Les taux de phosphates et de potassium doivent être contrôlés régulièrement et une supplémentation doit être mise en place si nécessaire. En cas d'hypophosphatémie de grade 3 persistant pendant ≥ 7 jours malgré un traitement de substitution maximal, OGSIVEO doit être suspendu jusqu'au retour à un grade ≤ 1 ou à la valeur initiale, puis il doit être repris à une dose de 100 mg deux fois par jour (voir rubrique Posologie et mode d'administration). En cas d'hypokaliémie de grade 3, quelle qu'en soit la durée, malgré un traitement de substitution maximal, OGSIVEO doit être suspendu jusqu'au retour à un grade ≤ 1 ou à la valeur initiale, puis il doit être repris à une dose de 100 mg deux fois par jour (voir rubrique Posologie et mode d'administration).
Anomalies hépatiques
Des augmentations des taux d'ALAT ou d'ASAT sont survenues chez les patients qui recevaient du nirogacestat (voir rubrique Effets indésirables). Des tests de la fonction hépatique doivent être effectués régulièrement. En cas d'ALT ou d'AST ≥3 à 5 x LSN, OGSIVEO doit être suspendu jusqu'à ce que l'ALT, l'AST ou les deux soient ramenés à <3 x LSN ou au niveau de base, puis il doit être repris à la dose de 100 mg deux fois par jour. En cas d'ALT ou d'AST > 5 x ULN, OGSIVEO doit être arrêté définitivement (voir section Posologie et mode d'administration).
Cancers cutanés non mélanomateux
Des cancers cutanés non mélanomateux (carcinome basocellulaire et carcinome épidermoïde) ont été rapportés chez des patients recevant du nirogacestat (voir rubrique Effets indésirables). Des examens cutanés doivent être effectués avant l'instauration du nirogacestat et régulièrement pendant le traitement par nirogacestat. Les cas doivent être pris en charge conformément aux pratiques cliniques et les patients peuvent poursuivre le traitement par le nirogacestat sans ajustement de la dose.
Toxicité embryo-foetale
OGSIVEO peut avoir des effets néfastes sur le foetus lorsqu'il est administré à une femme enceinte (voir rubriques Fertilité, grossesse et allaitement et Données de sécurité préclinique). Les patientes doivent être informées du risque potentiel pour le foetus. Les femmes en âge de procréer doivent avoir un test de grossesse négatif avant de commencer le traitement par OGSIVEO. Les femmes en âge de procréer recevant OGSIVEO doivent utiliser des méthodes contraceptives très efficaces pendant le traitement par OGSIVEO et pendant une semaine après la dernière dose d'OGSIVEO (voir rubrique Fertilité, grossesse et allaitement). Les femmes en âge de procréer doivent informer leur médecin en cas de grossesse avérée ou de suspicion de grossesse et d'arrêter de prendre le nirogacestat si elles débutent une grossesse.
Les patients de sexe masculin ayant des partenaires féminines en âge de procréer doivent utiliser une contraception hautement efficace pendant le traitement par OGSIVEO et pendant au moins 1 semaine après l'administration de la dernière dose de OGSIVEO (voir rubrique Fertilité, grossesse et allaitement).
Excipients
Ce médicament contient du lactose (voir rubriques 2 et Liste des excipients). Les patients présentant des troubles héréditaires rares d'intolérance au galactose, de déficit total en lactase ou de malabsorption du glucose- galactose ne doivent pas prendre ce médicament.
Ce médicament contient du jaune orangé S (E110) (voir rubriques 2 et Liste des excipients) qui peut provoquer des réactions allergiques.
Chaque comprimé pelliculé contient moins de 1 mmol (23 mg) de sodium, ce qui signifie qu'il est essentiellement sans sodium (voir rubrique Liste des excipients).
Résumé du profil de sécurité
Les données mentionnées ci-dessous reflètent l'exposition au nirogacestat chez 88 patients atteints de tumeurs desmoïdes dans 3 essais qui ont été traités par nirogacestat 150 mg deux fois par jour. La durée médiane d'exposition était de 21,5 mois.
L'effet indésirable grave le plus fréquemment rapporté était la toxicité ovarienne (ménopause précoce 3 %) qui était le seul effet indésirable grave rapporté par plus de 1 patiente dans les essais cliniques. Les effets indésirables graves les plus fréquents étaient la diarrhée (16 %) et l'hypophosphatémie (13%).
Un arrêt définitif dû à un effet indésirable est survenu chez 19 % des patients ayant reçu du nirogacestat. Les effets indésirables les plus fréquents ayant conduit à l'arrêt du traitement ont été la diarrhée (5 %), la toxicité ovarienne (5 %) et l'augmentation des ALAT (3 %).
Une interruption de traitement dû à un effet indésirable est survenu chez 59% des patients ayant reçu du nirogacestat. Les effets indésirables les plus fréquents ayant entraîné une interruption de traitement étaient la diarrhée (11 %), l'éruption maculo-papulaire (10 %), l'hypophosphatémie (6 %) et les nausées (5 %).
Une réduction de dose dû à un effet indésirable est survenu chez 44% des patients ayant reçu du nirogacestat. Les effets indésirables les plus fréquents ayant entraîné une réduction de la dose étaient la diarrhée (9 %), l'éruption maculo-papulaire (6 %), la stomatite (3 %) et l'hypophosphatémie (3 %).
Tableau récapitulatif des effets indésirables
Sauf indication contraire, les fréquences des effets indésirables sont basées sur les fréquences des effets indésirables toutes causes confondues identifiés chez 88 patients exposés au nirogacestat 150 mg deux fois par jour pendant une durée médiane de 21,5 mois dans les études cliniques.Les effets indésirables sont classés par ordre de fréquence selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 2 : Effets indésirables rapportés dans les études cliniques (N = 88)
Classe de système d'organes | Effet indésirable | Tous grades | Grades 3 à 4 |
Affections gastro- intestinales | Diarrhée | Très fréquent | Très fréquent |
Nausées | Très fréquent | Fréquent | |
Stomatite | Très fréquent | Fréquent | |
Sècheresse buccale | Très fréquent | -- | |
Affections de la peau et du tissu sous-cutané | Rasha | Très fréquent | Fréquent |
Alopécie | Très fréquent | -- | |
Folliculite | Très fréquent | Fréquent | |
Hidradénite | Fréquent | Fréquent | |
Sècheresse cutanée | Très fréquent | -- | |
Prurit | Très fréquent | -- | |
Tumeurs bénignes, malignes et non précisées | Carcinome basocellulaire | Fréquent | -- |
Carcinome épidermoïdeb | Fréquent | -- | |
Troubles du métabolisme et de la nutrition | Hypophosphatémie | Très fréquent | Très fréquent |
Hypokaliémie | Très fréquent | Fréquent | |
Affections du système nerveux | Céphalée | Très fréquent | -- |
| Vertige | Très fréquent |
|
Investigations | Protéinuriec | Très fréquent | -- |
Glycosuriec | Très fréquent | -- | |
Troubles du système sanguin et lymphatique | Eosinophilied | Très fréquent | -- |
Affections hépatobiliaires | Augmentation du taux des ALAT | Très fréquent | Fréquent |
Augmentation du taux des ASAT | Très fréquent | Fréquent |
Affections des organes de reproduction et du sein | Toxicité ovariennee | Très fréquent | -- |
Affections respiratoires, thoraciques et médiastinales | Toux | Très fréquent | -- |
Infection des voies respiratoires supérieuresf | Très fréquent | -- | |
Dyspnée | Très fréquent | -- | |
Épistaxis | Très fréquent | -- | |
Troubles généraux et anomalies au site d'administration | Fatigue | Très fréquent | Fréquent |
Maladie pseudo-grippale | Très fréquent | -- |
a Le terme « rash » comprend : rash maculopapuleux, dermatite acnéiforme, rash, rash érythémateux, rash prurigineux et rash papuleux.
b Le terme « carcinome épidermoïde » comprenait : carcinome épidermoïde cutané et carcinome épidermoïde.
c Les données de protéinurie et de glycosurie sont dérivées de résultats de paramètres biologiques anormaux qui se sont aggravés par rapport à l'entrée dans l'étude, et non d'événements indésirables rapportés.
d L'incidence de l'éosinophilie est basée sur les observations des valeurs de laboratoire plutôt que sur les ETEP rapportés.
e Le terme « toxicité ovarienne » comprend : insuffisance ovarienne, ménopause précoce, aménorrhée, oligoménorrhée, menstruation irrégulière, dysménorrhée, saignements menstruels abondants, sécheresse vulvo-vaginale, bouffées de chaleur, diminution de l'AMH et augmentation de la FSH. f Le terme « infection des voies respiratoires supérieures » comprend : IVRS, IVRS virale, sinusite aiguë et sinusite.
-- Signifie qu'aucun cas n'a été rapporté.
Description de certains effets indésirables
Les données mentionnées ci-dessous reflètent les résultats de l'étude randomisée de phase 3 NIR-DT- 301 menée chez des patients atteints de tumeurs desmoïdes, traités par 150 mg de nirogacestat (N = 69) ou un placebo (N = 72) deux fois par jour.
Diarrhée :
Dans la phase en double aveugle de l'étude NIR-DT-301, une diarrhée a été rapportée chez 84 % des patients recevant du nirogacestat contre 35 % des patients recevant le placebo. Des événements de grade 3 sont survenus chez 16 % et 1 % des patients, respectivement ; La diarrhée de grade ≤ 2 a disparu chez 74 % des patients qui ont poursuivi le traitement par le nirogacestat. Le délai médian d'apparition de la diarrhée chez les patients recevant le nirogacestat était de 9 jours (intervalle de 2 à 234 jours). La diarrhée a conduit à une réduction de la dose chez 10 % des patients et à l'arrêt du traitement chez 7 % des patients recevant le nirogacestat.
Affections de la peau et du tissu sous-cutané :
Dans la phase en double aveugle de l'étude NIR-DT-301, des réactions dermatologiques ont été rapportées à une incidence plus élevée chez les patients recevant du nirogacestat que chez ceux recevant le placebo ; elles comprenaient un rash maculopapuleux (32 % contre 6 %), une hidradénite (9 % contre 0) et une folliculite (13 % contre 0) (voir rubrique Mises en garde spéciales et précautions d'emploi). Le délai médian de survenue des éruptions cutanées était de 22 jours (intervalle de 2 à 603 jours). Les troubles cutanés et sous-cutanés ont conduit à une réduction de la dose chez 9 % des patients recevant le nirogacestat, y compris l'éruption maculo-papulaire chez 4 % et l'hidradénite chez 3 %. L'éruption maculo-papulaire a conduit à l'arrêt du traitement chez 1 % des patients.
Toxicité ovarienne :
Dans la phase en double aveugle de l'étude NIR-DT-301, 75 % des femmes en âge de procréer recevant du nirogacestat ont rapporté une toxicité ovarienne par rapport à aucune chez les patientes recevant le placebo. Trois effets indésirables graves de toxicité ovarienne ont été observés, tous liés à une ménopause prématurée.de toxicité ovarienne, toutes des ménopauses prématurées, représentant 11 % de l'ensemble des participantes rapportant une toxicité ovarienne. Le délai médian d'apparition de la toxicité ovarienne était de 8,9 semaines (de 1 jour à 54 semaines) et la durée médiane globale était de 18,9 semaines (de 11 jours à 215 semaines). La toxicité ovarienne a disparu chez 79% des femmes en âge de procréer pendant le traitement. Des informations de suivi sont disponibles pour l'ensemble des 27 patientes, à l'exception de deux d'entre elles ; après l'arrêt du traitement, la toxicité ovarienne a disparu chez toutes les femmes en âge de procréer pour lesquelles des données sont disponibles. Le délai médian de résolution après l'arrêt du nirogacestat était de 10,9 semaines (intervalle de 4 à 18 semaines).. Les effets du nirogacestat sur la fertilité sont inconnus (voir rubrique Mises en garde spéciales et précautions d'emploi). Une relation exposition-réponse a été identifiée entre le nirogacestat et les taux sériques d'hormone folliculostimulante (FSH), la FSH augmentant de manière linéaire avec l'augmentation des concentrations sériques de nirogacestat.
Anomalies électrolytiques :
Des anomalies électrolytiques sont survenues chez les patients recevant du nirogacestat dans la phase en double aveugle de l'étude NIR-DT-301, incluant une hypophosphatémie (43 %) et une hypokaliémie (12 %), par rapport à 7 % et 1 %, respectivement, chez les patients recevant le placebo. Le délai médian d'apparition de l'hypophosphatémie et de l'hypokaliémie était respectivement de 15 jours (de 1 à 833 jours) et de 15 jours (de 1 à 57 jours). Des cas d'hypophosphatémie et d'hypokaliémie de grade 3 sont survenus chez 3 % des patients recevant du nirogacestat, contre aucun chez les patients recevant le placebo (voir rubrique Mises en garde spéciales et précautions d'emploi). L'hypophosphatémie et l'hypokaliémie ont entraîné une réduction de la dose chez 4 % et 1 % des patients recevant le nirogacestat, respectivement. L'hypophosphatémie a conduit à l'arrêt de la dose chez 1% des patients recevant le nirogacestat.
Anomalies hépatiques :
Des augmentations des taux d'ALAT et d'ASAT sont survenues chez 19 % et 17 %, respectivement, des patients recevant du nirogacestat dans la phase en double aveugle de l'étude NIR-DT-301, contre 8 % et 11 %, respectivement, chez les patients recevant le placebo. Le délai médian d'apparition des premières élévations des ALAT et des ASAT était de 22 jours (ALAT : 8 à 924 jours ; ASAT : 1 à 1023 jours). Des élévations de grade 3 des ALAT et des ASAT (> 5 x LSN) sont survenues chez 3 % des patients traités par le nirogacestat contre 1 % dans le bras placebo (voir rubrique Mises en garde spéciales et précautions d'emploi). Les élévations des taux d'ALT et d'AST ont chacune conduit à une réduction de la dose chez 1 % des patients recevant le nirogacestat. Les élévations de l'ALT et de l'AST ont conduit à l'arrêt de la dose chez 4% et 3% des patients recevant le nirogacestat, respectivement.
Cancers cutanés non mélanomateux :
Des cancers cutanés non mélanomateux ont été signalés à une incidence plus élevée chez les patients recevant du nirogacestat que chez ceux recevant le placebo dans la phase en double aveugle de l'étude NIR-DT-301, notamment le carcinome épidermoïde (3 % contre 0) et le carcinome basocellulaire (1 % contre 0), un patient rapportant les deux types de cancer cutané non mélanomateux (voir rubrique Mises en garde spéciales et précautions d'emploi). Deux autres cas de cancer de la peau sans mélanome ont été signalés en dehors de la phase en double aveugle de l'étude NIR-DT-301.
Population pédiatrique
Des troubles épiphysaires, se manifestant par un élargissement de la plaque de croissance épiphysaire, ont été signalés chez 4 des 26 (15 %) patients pédiatriques présentant des plaques de croissance ouvertes et traités par le nirogacestat en dehors de l'étude NIR-DT-301. Les événements comprenaient l'épiphysiolyse, la fracture de la hanche, le trouble épiphysaire et l'ostéonécrose. Les quatre patients pédiatriques étaient âgés de 11 à 12 ans. Voir la rubrique Posologie et mode d'administration pour des informations sur l'utilisation pédiatrique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté selon les modalités définies dans le Protocole d'utilisation thérapeutique et de recueil de données (voir PUT RD).
SURVEILLANCE :
- Les taux de phosphates et de potassium doivent être contrôlés régulièrement.
- Des tests de la fonction hépatique doivent être effectués régulièrement.
- Des examens cutanés doivent être effectués avant l'instauration du nirogacestat et régulièrement pendant le traitement.
Avant d'entamer le traitement, INFORMER les patientes en âge de procréer du risque de toxicité ovarienne.
La
contraception doit être utilisée pendant le traitement et une semaine après la
dernière dose.
Le
don de gamètes ne peut être fait pendant le traitement et une semaine après la
dernière dose.
Femmes en âge de procréer/Contraception chez les hommes et les femmes
Les femmes en âge de procréer et les hommes ayant des partenaires féminines en âge de procréer doivent être informés que toute grossesse doit être évitée pendant le traitement par OGSIVEO (voir rubrique Mises en garde spéciales et précautions d'emploi).
Les femmes en âge de procréer recevant OGSIVEOdoivent utiliser des méthodes de contraception hautement efficaces pendant le traitement par OGSIVEOet pendant 1 semaine après l'administration de la dernière dose de OGSIVEO (voir rubrique Mises en garde spéciales et précautions d'emploi). On ne sait pas si OGSIVEOpeut réduire l'efficacité des contraceptifs hormonaux à action systémique.Par conséquent, il convient de conseiller aux patientes utilisant des contraceptifs hormonaux d'utiliser un autre moyen de contraception non affecté par les inducteurs enzymatiques (par exemple, un dispositif intra-utérin non hormonal) ou un autre moyen de contraception non hormonal (par exemple, un préservatif [avec spermicide, le cas échéant]) pendant le traitement par OGSIVEO et pendant au moins une semaine après la dernière dose de OGSIVEO.
Les femmes en âge de procréer doivent informer leur médecin en cas de grossesse avérée ou de suspicion de grossesse et d'arrêter de prendre le nirogacestat si elles débutent une grossesse. Les femmes en âge de procréer ne doivent pas donner d'ovules (ovocytes) pendant qu'elles prennent OGSIVEO et pendant une semaine après avoir reçu la dernière dose de OGSIVEO.
Les patients de sexe masculin ayant des partenaires féminines en âge de procréer doivent utiliser une contraception hautement efficace pendant le traitement par nirogacestat et pendant 1 semaine après l'administration de la dernière dose de nirogacestat (voir rubrique Mises en garde spéciales et précautions d'emploi). Les patients de sexe masculin ne doivent pas faire de don de sperme pendant le traitement et pendant une semaine après avoir reçu la dernière dose de OGSIVEO.
Grossesse
Il existe des données limitées sur l'utilisation de OGSIVEO chez la femme enceinte. D'après les résultats d'études menées chez l'animal et son mécanisme d'action, OGSIVEO est susceptible de provoquer des malformations foetales lorsqu'il est administré à une femme enceinte (voir rubrique Données de sécurité préclinique). Une absence de grossesse chez les femmes en âge de procréer doit être vérifiée 7 jours avant de commencer le traitement par OGSIVEO. Les patientes doivent être informées du risque potentiel pour le foetus et OGSIVEO ne doit pas être utilisé pendant la grossesse. Si une patiente débute une grossesse pendant le traitement par OGSIVEO, le traitement doit être interrompu. Un avortement spontané a été rapporté par une femme de l'étude DeFi qui a conçu un enfant alors qu'elle recevait du nirogacestat.
Allaitement
Il n'existe pas de données sur l'excrétion du nirogacestat ou de ses métabolites dans le lait maternel chez l'homme ou l'animal, ou sur ses effets chez les nourrissons allaités ou sur la production de lait. En raison du risque d'effets indésirables graves chez un nourrisson allaité, Les femmes ne doivent pas allaiter pendant le traitement par OGSIVEO et pendant 1 semaine après l'administration de la dernière dose de OGSIVEO (voir rubrique Contre-indications).
Fertilité
Aucune étude de fertilité n'a été menée chez l'humain. L'effet de OGSIVEO sur la fertilité chez l'humain n'est pas connu. D'après les résultats des études effectuées chez l'animal, la fertilité des mâles et des femelles peut être altérée (voir rubrique Données de sécurité préclinique).
Les études d'interaction n'ont été réalisées que chez l'adulte.
Le nirogacestat est principalement métabolisé par le CYP3A4 et est un substrat de la glycoprotéine P (P-gp) in vitro.
Agents susceptibles d'augmenter les concentrations sériques de nirogacestat
Effet des inhibiteurs modérés et puissants du CYP3A4
Dans une étude clinique, l'administration concomitante d'itraconazole (un puissant inhibiteur du CYP3A4 et de la P-gp) a augmenté la Cmax du nirogacestat de 2,5 fois et l'ASC de 8,2 fois. L'administration concomitante d'inhibiteurs modérés du CYP3A4 devrait également entraîner une augmentation de l'exposition cliniquement pertinente.
Un traitement concomitant avec des inhibiteurs puissants du CYP3A4 (clarithromycine, , ketaconazole oral, itraconazole) et des inhibiteurs modérés du CYP3A4 (erythromycyine, fluconazole) doit donc être évité..
Il convient d'envisager d'autres médicaments concomitants ne présentant pas d'inhibition ou d'induction du CYP3A4 ou présentant une inhibition ou une induction minimale. Si aucune alternative thérapeutique n'est disponible, OGSIVEO doit être immédiatement interrompu pendant la période où un inhibiteur puissant ou modéré du CYP3A4est administré.
Les patients doivent éviter de consommer du pamplemousse et du jus de pamplemousse lors de la prise de OGSIVEO car ils contiennent des inhibiteurs du CYP3A4. (voir rubrique Posologie et mode d'administration).
Agents susceptibles de diminuer les concentrations sériques de nirogacestat
Effet des inducteurs puissants et modérés du CYP3A4
Les effets des inducteurs du CYP3A4 sur l'exposition au nirogacestat n'ont pas été évalués dans une étude clinique. On s'attend à ce que les inducteurs modérés et forts entraînent des diminutions cliniquement pertinentes de l'exposition au nirogacestat qui pourraient conduire à une réduction de l'efficacité. Un traitement concomitant par des inducteurs puissants du CYP3A4 (p. ex. carbamazépine, phénytoïne, rifampicine, phénobarbital et millepertuis) et des inducteurs modérés du CYP3A (p. ex. éfavirenz et étravirine) doit donc être évité. Chez les patients pour lesquels des inducteurs du CYP3A4 sont indiqués, d'autres médicaments ayant un potentiel d'induction enzymatique moindre doivent être choisis. Si aucune alternative thérapeutique n'est disponible, OGSIVEO doit être interrompu pendant la période où un inducteur modéré ou puissant du CYP3A4 est administré en raison d'un risque de perte d'efficacité .
Effet des agents réducteurs de la sécrétion d'acide gastrique
Le nirogacestat a une solubilité dépendante du pH, avec une solubilité considérablement réduite à un pH supérieur à 6,0. Les effets des médicaments diminuant la sécrétion d'acide gastrique (antagonistes des récepteurs H2 et inhibiteurs de la pompe à protons et antiacides) sur l'exposition au nirogacestat n'ont pas été évalués dans une étude clinique ; cependant, l'administration concomitante de ces médicaments avec le nirogacestat peut réduire la biodisponibilité du nirogacestat. Éviter l'utilisation concomitante avec les inhibiteurs de la pompe à protons et les bloqueurs H2. Si l'utilisation concomitante ne peut être évitée, le nirogacestat peut être administré en décalage avec les antiacides en administrant OGSIVEO 2 heures avant ou 2 heures après la prise d'un antiacide.
Effets du nirogacestat sur la pharmacocinétique d'autres médicaments
Substrats sélectifs des isoformes du CYP
Une étude d'interaction médicamenteuse menée chez des volontaires sains et portant sur les effets de doses multiples de nirogacestat à raison de 95 mg une fois par jour sur l'exposition au midazolam, un substrat sensible du CYP3A4, a entraîné une multiplication par 1,3 de la Cmax du midazolam et une multiplication par 1,6 de l'aire sous la courbe du midazolam. L'effet de la dose clinique de nirogacestat
(150 mg deux fois par jour) sur l'exposition au midazolam n'a pas été étudié et pourrait être différent. OGSIVEO ne doit pas être utilisé en cas d'administration concomitante de substrats du CYP3A4 ayant des indices thérapeutiques étroits (par exemple, ciclosporine, tacrolimus, digitoxine, warfarine, carbamazépine).
Des études in vitro ont montré que le nirogacestat peut inhiber le CYP3A4 et induire le CYP2C8, CYP2C9, CYP2C19 et le CYP2B6 et il existe donc un risque que OGSIVEO entraîne une diminution de l'exposition des substrats de ces enzymes.Lorsque des substrats de CYP2C8, CYP2C9, CYP2C19 et CYP2B6 sont administrés avec OGSIVEO, une surveillance des effets indésirables et une évaluation de l'efficacité réduite du substrat doivent être effectuées et un ajustement de la dose du substrat peut être nécessaire pour maintenir des concentrations plasmatiques optimales.
Aucune étude n'ayant été réalisée sur l'effet du nirogacestat sur l'exposition aux stéroïdes contraceptifs systémiques, on ne sait pas si le nirogacestat réduit l'efficacité des contraceptifs hormonaux à action systémique. Les femmes en âge de procréer doivent utiliser des méthodes contraceptives très efficaces (voir rubrique Fertilité, grossesse et allaitement).
Systèmes de transport de médicaments
Une étude d'interaction médicamenteuse à dose unique a montré que le nirogacestat n'affectait pas l'exposition au dabigatran, un substrat de la P-gp, ce qui confirme l'absence d'inhibition cliniquement significative de la P-gp par le nirogacestat.
Inhibiteurs de la protéine de résistance du cancer du sein (BCRP)
On ne sait pas si le nirogacestat est un substrat de la BCRP. OGSIVEO ne doit pas être utilisé en cas d'administration concomitante d'inhibiteurs de la BCRP (par exemple, ciclosporine, darolutamide, fostamatinib).
Le traitement par OGSIVEO doit être instauré par un médecin expérimenté dans l'utilisation des médicaments anticancéreux
Posologie
La dose recommandée est de 150 mg par voie orale deux fois par jour. Cette dose ne doit pas être dépassée.
Durée du traitement
OGSIVEO doit être poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable
Dose oubliée
En cas d'oubli d'une dose de OGSIVEO, les patients ne doivent pas prendre de dose supplémentaire. Les patients doivent prendre la dose suivante prescrite.
Ajustements posologiques en cas d'effets indésirables
Les modifications posologiques recommandées pour certains effets indésirables sévères sont présentées dans le Tableau 1 ci-dessous.
Pour les autres effets indésirables graves, ou en cas d'effets indésirables mettant en jeu le pronostic vital, OGSIVEO doit être suspendu jusqu'à ce que l'effet soit revenu à un grade ≤ 1 ou à l'état initial. OGSIVEO ne doit être repris qu'à une dose de 100 mg deux fois par jour et uniquement après avoir soigneusement examiné le bénéfice potentiel et la probabilité de réapparition de l'effet indésirable. OGSIVEO doit être définitivement arrêté en cas de réapparition d'un effet indésirable grave ou mettant en jeu le pronostic vital lors de la reprise du traitement à la dose réduite.
Des modifications posologiques doivent être effectuées si les patients présentent les effets indésirables suivants (les grades font référence aux critères communs de terminologie pour les événements indésirables) :
Tableau 1 : Modifications posologiques recommandées en cas d'effets indésirables chez les patients traités par nirogacestat
Effet indésirable | Action recommandée |
Diarrhée | |
Diarrhée de grade 3 persistant pendant ≥ 3 jours malgré un traitement médical maximal | OGSIVEO doit être suspendu jusqu'au retour de l'effet à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour. |
Réactions cutanées | |
Folliculite de grade 3 | OGSIVEO doit être suspendu jusqu'au retour de l'effet à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour. |
Rash maculopapuleux de grade 3 | OGSIVEO doit être suspendu jusqu'au retour de l'effet à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour. |
Hidradénite de grade 3 | OGSIVEO doit être suspendu jusqu'au retour de l'effet à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour. |
Anomalies électrolytiques | |
Hypophosphatémie de grade 3 persistant pendant ≥ 7 jours malgré un traitement de substitution maximal | OGSIVEO doit être suspendu jusqu'au retour de l'effet à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour. |
Hypokaliémie de grade 3 malgré un traitement de substitution maximal | OGSIVEO doit être suspendu jusqu'au retour de l'effet à un grade ≤ 1 ou à l'état initial, puis il doit être repris à une dose de 100 mg deux fois par jour. |
Effet indésirable | Action recommandée |
Anomalies hépatiques | |
Taux d'ALAT ou d'ASAT ≥ 3 à 5 x LSN | OGSIVEO doit être suspendu jusqu'à ce que les taux d'ALAT, d'ASAT ou les deux soient revenus à un taux < 3 x LSN ou à la valeur initiale, puis il doit être repris à une dose de 100 mg deux fois par jour. |
Taux d'ALAT ou d'ASAT > 5 x LSN | OGSIVEO doit être définitivement arrêté. |
Autres effets indésirables | |
Anaphylaxie ou autre réaction d'hypersensibilité sévère | OGSIVEO doit être définitivement arrêté. |
Populations particulières
Personnes âgées
Aucun ajustement posologique n'est recommandé chez les patients âgés de 65 ans ou plus. Les données cliniques chez les patients agés de plus de 65 ans sont limités.
Insuffisance rénale
Aucun ajustement posologique n'est recommandé chez les patients insuffisants rénaux légers à modérés (voir rubrique Propriétés pharmacocinétiques).
OGSIVEO n'est pas recommandé chez les patients atteints d'insuffisance rénale sévère.
Insuffisance hépatique
Aucun ajustement posologique n'est recommandé chez les patients présentant une insuffisance hépatique légère ou modérée. L'administration de OGSIVEO n'est pas recommandée chez les patients présentant une insuffisance hépatique sévère (voir section Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité d'emploi et l'efficacité du nirogacestat chez les patients âgés de moins de 18 ans n'ont pas été établies. OGSIVEO ne doit pas être utilisé chez les enfants de la naissance à moins de 2 ans en raisonde problèmes de sécurité potentiels liés à la croissance structurelle et fonctionnelle. . Les données actuellement disponibles sont décrites aux rubriques Effets indésirables et Propriétés pharmacodynamiques, mais aucune recommandation sur la posologie ne peut être donnée
Mode d'administration
OGSIVEO est destiné à être administré par voie orale.
Les comprimés peuvent être pris pendant ou en dehors des repas et ne doivent pas être mâchés ou écrasés. Les patients doivent éviter de consommer du pamplemousse ou du jus de pamplemousse pendant qu'ils prennent du nirogacestat. (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Durée de conservation :
5 ans
Précautions particulières de conservation :Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Signes et symptômes
L'expérience avec des doses supérieures à la dose thérapeutique recommandée est limitée. Les symptômes d'un surdosage de OGSIVEO devraient être une extension de ses actions pharmacologiques et peuvent inclure diarrhée, nausées, vomissements, hypophosphatémie, augmentation du taux des transaminases et épistaxis.
Prise en charge du surdosage
En raison du taux élevé de liaison aux protéines, OGSIVEO ne devrait pas être dialysable chez les patients présentant des taux normaux de protéines sériques. En cas de surdosage, le traitement par OGSIVEO doit être arrêté et un traitement symptomatique doit être initié
Classe pharmacothérapeutique : agent antinéoplasique ; Code ATC : L01XX81
Mécanisme d'action
Le nirogacestat est un inhibiteur oral, sélectif, réversible et non compétitif de la gamma-sécrétase qui bloque l'activation protéolytique des récepteurs Notch et d'autres récepteurs. En cas de dérèglement, les récepteurs Notch peuvent jouer un rôle dans l'activation des voies qui contribuent à la croissance tumorale.
Électrophysiologie cardiaque
Les effets de la concentration de nirogacestat sur l'allongement de l'intervalle QTc ont été prédits à l'aide d'une analyse basée sur un modèle. Les intervalles de confiance à 90 % pour le changement moyen prévu de l'intervalle QTcF étaient inférieurs à 10 msec à la Cmax prévue à des doses suprathérapeutiques. Par conséquent, aucun allongement cliniquement significatif de l'intervalle QTcF n'est associé au dosage thérapeutique du nirogacestat.
Efficacité et sécurité cliniques
L'étude NIR_DT-301 DeFi était une étude de phase 3 internationale, multicentrique, randomisée (1:1), en double aveugle, contrôlée contre placebo, menée chez des patients adultes atteints de tumeurs desmoïdes en progression selon les critères RECIST v1.1. Les patients atteints de tumeurs desmoïdes confirmées histologiquement et ayant progressé de ≥ 20 % selon la méthode RECIST v1.1 dans les 12 mois suivant la sélection et les tumeurs desmoïdes dont la progression continue n'entraîne pas de risque significatif immédiat pour le patient sont éligibles. La randomisation a été stratifiée par localisation(s) de la tumeur cible (intra-abdominale ou extra-abdominale). Les patients présentant plusieurs tumeurs cibles situées à la fois dans la zone intra-abdominale et extra-abdominale ont été classés dans le groupe tumeur intra-abdominale. Les patients ont reçu 150 mg de nirogacestat ou un placebo par voie orale, deux fois par jour, jusqu'à la progression de la maladie, le décès ou le retrait de l'étude pour une autre raison. La mesure principale de l'efficacité était la survie sans progression (SSP). La progression a été déterminée par radiographie à l'aide des critères RECIST v1.1 par un examen d'imagerie central indépendant en aveugle, ou évaluée cliniquement par l'investigateur et qualifié par un examen central indépendant en aveugle, ou par le décès toutes causes confondues. Les autres mesures de l'efficacité comprenaient le taux de réponse objective (TRO), la variation par rapport à l'entrée dans l'étude de la douleur au Cycle 10, la variation par rapport à l'entrée dans l'étude de la sévérité des symptômes spécifiques de la tumeur desmoïde au Cycle 10, la variation par rapport à l'entrée dans l'étude de la fonction de rôle et du fonctionnement physique au Cycle 10, et la variation par rapport à l'entrée dans l'étude de la qualité de vie globale au Cycle 10. La douleur a été mesurée par la moyenne sur 7 jours de l'item n° 3 (c'est-à-dire la pire douleur) de l'inventaire abrégé de la douleur (Brief Pain Inventory, BPI). La gravité des symptômes spécifiques de la tumeur desmoïde et le fonctionnement physique ont été mesurés à l'aide de l'échelle d'évaluation des symptômes/de l'impact des tumeurs desmoïdes (GOunder/DTRF DEsmoid Symptom/Impact Scale, GODDESS). La fonction de rôle, le fonctionnement physique et la qualité de vie globale ont été mesurés par le questionnaire sur la qualité de vie à 30 items de l'Organisation européenne pour la recherche et le traitement du cancer (European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-Core 30, EORTC QLQ-C30).
Au total, 142 patients ont été randomisés : 70 dans le groupe nirogacestat et 72 dans le groupe placebo. Globalement, l'âge médian était de 34 ans (intervalle : 18 à 76 ans) ; 65 % étaient des femmes ; l'origine ethnique était la suivante : 83 % de Caucasiens, 6 % de Noirs, 3 % d'Asiatiques et 8 % d'autres origines ; 73 % avaient un indice de performance (IP) ECOG de 0, 27 % avaient un IP ECOG de 1, et moins de 1 % avaient un IP ECOG de 2. Vingt-trois pour cent des patients présentaient une maladie intra- abdominale ou une maladie intra-abdominale et extra-abdominale, et 77 % présentaient uniquement une maladie extra-abdominale. Quarante-et-un pour cent des patients souffraient d'une maladie multifocale et 59 % d'une maladie focale unique. Sur 105 patients avec un statut de mutation somatique tumorale connu, 81 % étaient porteurs d'une mutation du gène CTNNB1 et 21 % d'une mutation du gène APC. Dix-sept pour cent des patients avaient des antécédents familiaux de polypose adénomateuse familiale (PAF). Vingt-trois pour cent des patients n'avaient reçu aucun traitement antérieur et 44 % avaient reçu ≥ 3 lignes de traitement antérieures. Le traitement antérieur comprenait une intervention chirurgicale (53 %), une radiothérapie (23 %) et un traitement systémique (61 %). Trente-trois pour cent des patients avaient déjà été traités par un inhibiteur de tyrosine kinase et 36 % avaient déjà été traités par chimiothérapie. Cinquante pour cent avaient un score BPI-SF ≥ 2 pour l'item 3 (pire douleur) à l'entrée dans l'étude.
Les résultats d'efficacité de la population ITT, qui comprenait tous les patients randomisés, sont présentés ci-dessous. Les améliorations de la SSP et du TRO étaient en faveur du nirogacestat, indépendamment des caractéristiques initiales, incluant la localisation de la tumeur et le type de traitements antérieurs.
Tableau 3 : Résultats d'efficacité chez les patients présentant des tumeurs desmoïdes en progression selon les critères RECIST 1.1
| Nirogacestat N = 70 | Placebo N = 72 |
Survie sans progression | ||
Nombre (%) de patients présentant un événement | 12 (17) | 37 (51) |
Progression radiographiquea | 11 (16) | 30 (42) |
Progression cliniquea | 1 (1) | 6 (8) |
Décès | 0 | 1 (1) |
Médiane (mois) (IC à 95 %)b | NA (NA, NA) | 15,1 (8,4 ; NA) |
Risque relatif (IC à 95 %) | 0,29 (0,15 ; 0,55) | |
Valeur pc | < 0,001 | |
Taux de réponse objectiveErreur ! Source du renvoi introuvable. | ||
TRO, n (%) IC à 95 % d | 29 (41) (29,8 ; 53,8) | 6 (8) (3,1 ; 17,3) |
RC | 5 (7) | 0 |
RP | 24 (34) | 6 (8) |
Valeur pe | < 0,001 | |
Abréviations : IC : intervalle de confiance ; NA : non atteint ; RC : réponse complète ; RP : réponse partielle ; TRO : taux de réponse objective
Erreur ! Source du renvoi introuvable.Évalué par un examen central indépendant en aveugle.
b Obtenu à l'aide de la méthodologie de Kaplan-Meier.
c La valeur p était issue d'un test du log-rank stratifié unilatéral avec le placebo comme référence.
d Obtenu en utilisant une méthode exacte basée sur la distribution binomiale.
e La valeur p était issue d'un test bilatéral de Cochran-Mantel-Haenszel.
Figure 1 : Courbe de Kaplan-Meier de la SSP
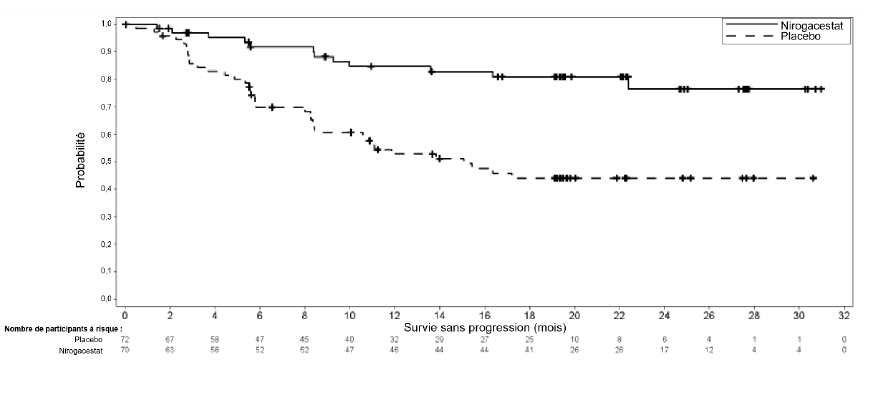
Remarque : les intervalles de confiance médians et à 95 % ont été estimés à partir de la méthode de Kaplan-Meier. En raison du faible nombre d'événements dans le bras nirogacestat, l'estimation de Kaplan-Meier du délai médian jusqu'à la progression n'a pas pu être effectuée.
Comparativement au placebo au Cycle 10, les patients du bras nirogacestat ont rapporté une réduction statistiquement significative de la douleur, évaluée par le questionnaire BPI-SF (-1,583 versus -0,241 ; différence moyenne des MC = -1,342 ; p < 0,001), ainsi qu'une amélioration statistiquement significative, par rapport à l'entrée dans l'étude, du score total des symptômes de l'échelle GODDESS DTSS (-1,110 versus 0,457 ; différence de la moyenne des MC = -1,567 ; p < 0,001), du score du domaine du fonctionnement physique de l'échelle GODDESS DTIS (-0,613 versus 0,094 ; différence de la moyenne des MC = -0,706 ; p < 0,001). Les patients du bras nirogacestat ont également rapporté une amélioration statistiquement et cliniquement significative par rapport à l'entrée dans l'étude au Cycle 10 par rapport au placebo pour les évaluations des RRP des scores du questionnaire sur l'état de santé global/la QdV de l'EORTC (2,935 versus ‐8,466 ; différence de la moyenne des MC = 11,401 ; p = 0,006, fonctionnement physique selon le questionnaire EORTC QLQ-C30 (9,143 versus - 5,225 ; différence de la moyenne des MC = 14,368 ; p < 0,001 ; p<0,001), fonction de rôle du questionnaire EORTC QLQ- C30 (13,293 versus -5,590 ; différence de la moyenne des MC = 18,884 ; p < 0,001). Par rapport au placebo, les patients du bras nirogacestat ont rapporté des améliorations précoces et durables de la douleur, du fonctionnement physique, de la fonction de rôle et des symptômes de la tumeur desmoïde.
Population pédiatrique
La sécurité d'emploi et l'efficacité de OGSIVEO chez les enfants n'ont pas été établies.
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec le nirogacestat dans tous les sous-groupes de la population pédiatrique âgée de plus de 2 ans.
Absorption
Les concentrations maximales de nirogacestat sont atteintes environ 1 heure après l'administration par voie orale. La biodisponibilité du nirogacestat après administration par voie orale est d'environ 19,2 % (intervalle : 16,2 % - 24,3 %). Une comparaison des valeurs de Cmax et d'ASC normalisées en fonction de la dose chez les patients atteints de tumeurs solides avancées indique une diminution de 6,7 % (rapport des moyennes géométriques normalisées en fonction de la dose [dn-GMR] et IC à 90 % : 93,32% [54,6 % - 162,44 %]) de la Cmax et une augmentation de 14 % (dn-GMR et IC à 90 % : 114,03 % [76,2 % - 170,63 %]) de l'ASC lorsque le nirogacestat est administré avec un repas riche en matières grasses par rapport à une administration à jeun. La variabilité de ces évaluations est élevée.
Distribution
Le rapport sang/plasma du nirogacestat est estimé à 0,52 chez l'humain. La liaison aux protéines sériques est d'environ 99,6 % in vitro. Le nirogacestat est fortement lié à la fois à l'albumine sérique humaine et à l'α-1 glycoprotéine acide, mais avec une plus grande affinité pour l'α-1 glycoprotéine acide. D'après l'analyse pharmacocinétique de la population, le volume oral apparent du compartiment central de distribution du nirogacestat chez les patients atteints de tumeurs desmoïdes a été estimé à 1 430L
Biotransformation
La principale voie métabolique du nirogacestat est la N-désalkylation via le CYP3A4. Le nirogacestat est le principal composant circulant dans le sérum. Le nirogacestat est largement métabolisé. De nombreux métabolites mineurs ont été détectés dans la circulation et les excréments. Les données in vitro n'ont pas permis d'identifier de métabolites majeurs ou actifs. Cependant, en raison des limites de la détection des métabolites non marqués, les connaissances sur les métabolites majeurs ou actifs in vivo sont incomplètes.
Élimination
Après l'administration d'une dose orale unique de nirogacestat radiomarqué chez des sujets sains, environ 65 % de la dose sont récupérés au cours des 13 jours suivant l'administration ; 38,1 % sont éliminés dans les fèces, 17 % sont éliminés dans l'urine et 9,7 % du produit radiomarqué récupéré sont retrouvés dans l'air expiré. Le nirogacestat sous forme inchangée dans les urines représente moins de 0,01 % de la dose administrée.
Dans une population atteinte de tumeurs solides avancées, la demi-vie d'élimination terminale apparente est d'environ 25,3 heures. La clairance systémique apparente est d'environ 23,4 l/h.
Linéarité/non-linéarité
L'exposition au nirogacestat augmente avec l'augmentation des doses uniques et répétées, avec des augmentations proportionnelles dans l'intervalle des doses de 50 à 150 mg.
Les concentrations à l'état d'équilibre sont atteintes environ 7 jours après l'administration répétée. L'analyse pharmacocinétique de population estime un ratio d'accumulation d'environ 1,53 chez les patients atteints de tumeurs desmoïdes.
Populations particulières
Effets de l'insuffisance hépatique
La pharmacocinétique du nirogacestat a été évaluée chez des patients atteints d'insuffisance hépatique modérée à sévère selon la classification de Child-Pugh. Chez les patients atteints d'insuffisance hépatique modérée, l'exposition totale au nirogacestat (ASC) n'a pas été modifiée par l'insuffisance hépatique modérée, mais l'exposition maximale (Cmax) a été réduite de 28 % en raison d'un volume de distribution plus élevé et d'une demi-vie plus longue.
Effets de l'insuffisance rénale
Les effets de l'insuffisance rénale sur la pharmacocinétique du nirogacestat n'ont pas été évalués dans un essai clinique spécifique. Dans un modèle PopPK, aucune relation cliniquement significative n'a été observée entre les tests de la fonction rénale et la pharmacocinétique du nirogacestat. Sur les 335 sujets inclus dans l'analyse PopPK, deux présentaient une insuffisance rénale légère et une insuffisance rénale modérée. Aucun sujet présentant une insuffisance rénale sévère n'a été inclus dans l'analyse PopPK.
Le nirogacestat n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. La fatigue et les vertiges pouvant survenir chez certains patients prenant du nirogacestat (voir rubrique Effets indésirables), les patients qui présentent ces effets indésirables doivent être prudents lorsqu'ils conduisent ou utilisent des machines.
Dans les études de toxicité à doses répétées chez le rat et le chien, la plupart des toxicités ont été associées à l'inhibition de la gamma-sécrétase. Les effets comprenaient une atrophie ovarienne, des altérations du cycle oestral, une diminution de la cellularité du tissu lymphoïde associé à l'intestin et une diminution de la cellularité des ganglions lymphatiques mésentériques. Dans l'étude sur les rats, on a observé un épaississement de la plaque de croissance, mais limité aux os en croissance. En outre, toutes les doses évaluées dans l'étude sur les rats ont montré une néphropathie chronique progressive,
une phospholipidose pulmonaire et une nécrose des glandes salivaires de manière dose-dépendante. Dans l'étude sur le chien, des effets liés au traitement ont été observés dans les intestins, la rate, la vésicule biliaire, le foie, les reins, les testicules et les ovaires. Les résultats concernant les intestins et le foie étaient associés à une inflammation généralisée et à des changements pathologiques cliniques connexes chez la plupart des chiens. Aucun NOAEL n'a été identifié dans les études de toxicité orale de 3 mois chez les rats et les chiens. La dose la plus faible dans l'étude sur les rats était de 5 mg/kg/jour (dose équivalente chez l'homme de 50 mg/jour) et chez les chiens, la dose la plus faible était de 2 mg/kg/jour (dose équivalente chez l'homme de 70 mg/jour). Les expositions systémiques étaient également inférieures aux expositions systémiques humaines (AUC) après administration de 150 mg BID de nirogacestat.
Carcinogénicité
La signalisation Notch semble avoir une fonction à la fois oncogène et suppressive de tumeur. Le potentiel carcinogène du nirogacestat a été évalué dans une étude de 6 mois chez des souris rasH2 transgéniques. À des doses allant jusqu'à 100 mg/kg/jour, aucune augmentation significative de la tumorigénicité n'a été observée. À la dose de 100 mg/kg/jour, les expositions systémiques (ASC) étaient inférieures (0,2 fois) à celles des humains ayant reçu 150 mg de nirogacestat 2x/j. Le potentiel carcinogène chez le rat n'a pas été évalué.
Toxicité pour la reproduction et le développement
Le nirogacestat a réduit les indices de fertilité chez les rats mâles et femelles, ce qui était corrélé à l'atrophie ovarienne, à une réduction du poids des testicules et à une diminution de la motilité des spermatozoïdes et les effets sur la morphologie des spermatozoïdes. En outre, une perte embryonnaire précoce est survenue dans les études de fertilité. Dans une étude préliminaire sur le développement embryo-foetal, le nirogacestat a induit une perte d'embryon significative et liée à la dose, des résorptions précoces et une diminution du poids foetal chez les embryons survivants. Ces effets sont survenus à 20 mg/kg/jour, entraînant des expositions systémiques inférieures (environ 0,45 fois) à celles de l'humain après l'administration d'une dose de 150 mg de nirogacestat 2x/j (voir rubrique Mises en garde spéciales et précautions d'emploi).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
OGSIVEO 100 mg, comprimés pelliculés
Comprimés pelliculés ronds, de couleur orange clair, de 10 mm de diamètre, portant la mention « 100 » gravée sur une face.
OGSIVEO 100 mg, comprimés pelliculés
Flacon en PEHD avec fermeture de sécurité enfant, scellé par induction, contenant 75 comprimés.
OGSIVEO 100 mg, comprimés pelliculés
Chaque comprimé pelliculé contient 100 mg de nirogacestat (sous forme de dihydrobromure de nirogacestat).
Excipients à effet notoire
Chaque comprimé pelliculé contient 115,7 mg de lactose monohydraté.
Chaque comprimé pelliculé contient du jaune orangé S (E110).
Pour la liste complète des excipients, voir rubrique Liste des excipients
Noyau du comprimé :
Cellulose microcristalline
Lactose monohydraté
Glycolate d'amidon sodique, type A
Stéarate de magnésium
Pelliculage :
Opadry® QX orange 321A130059-CN (comprimés de 100 mg)
L'Opadry® QX orange contient :
Copolymère greffé d'alcool polyvinylique et de macrogol (PEG) (E1209)
Talc (E553b)
Dioxyde de titane (E171)
Monocaprylocaprate de glycérol type 1/mono/diglycérides (E471)
Alcool polyvinylique - partiellement hydrolysé (E1203)
Laque aluminique de FD&C jaune n° 6/jaune orangé S (E110)
Oxyde de fer jaune (E172)